Spectroscopie
1)Généralités
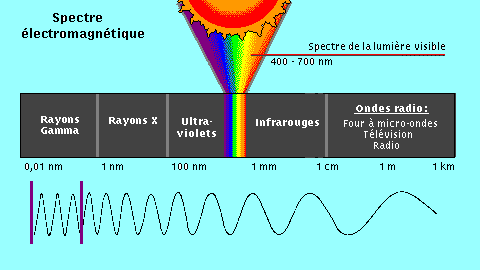
2)Infra-rouge
L'IR concerne le nombre et le type de liaisons chimiques. Son domaine s'étend de 4000 à 400 cm-1.
Chaque liaison d'une molécule vibre en permanence à une fréquence qui dépend:
 du type d'atomes de la liaison
du type de la liaison.
Seules les vibrations qui font varier le moment dipolaire de la molécule
absorbent les radiations infrarouges.Et donc les molécules qui n'absorbent
pas dans l'IR sont celles qui n'ont pas de moment dipolaire permanent.
Par ailleurs,dans la zone du proche infrarouge, les absorptions ne sont pas dues
aux vibrations fondamentales des molécules, mais aux vibrations harmoniques et
aux vibrations de combinaisons.
du type d'atomes de la liaison
du type de la liaison.
Seules les vibrations qui font varier le moment dipolaire de la molécule
absorbent les radiations infrarouges.Et donc les molécules qui n'absorbent
pas dans l'IR sont celles qui n'ont pas de moment dipolaire permanent.
Par ailleurs,dans la zone du proche infrarouge, les absorptions ne sont pas dues
aux vibrations fondamentales des molécules, mais aux vibrations harmoniques et
aux vibrations de combinaisons.
Chaque pic d'absorption est donc caractéristique d'un certain type de liaison et on distingue
les vibrations de stretching, généralement intenses de 4000-2000cm-1
les vibrations de bending de 2000-1500 cm-1
la region de 1500-600 cm-1 étant en général qualifiée d'empreinte digitale.

Exemples
| Mode de Vibration | Groupe Fonctionnel | Bande d'absorption(cm-1) |
| Stretching | O-H | 3590-3650 |
| Stretching | C-H | 2850-3300 |
| Stretching | C-O | 900-1200 |
| Stretching | C-Cl | 700 |
| Stretching | C-F | 1100 |
| Stretching | C=C | 1600-1680 |
| Stretching | C=O | 1700-1750 |
| Stretching | C=-C | 2000-2200 |
| Stretching | C=-N | 2200-2300 |
| Bending | -CH2 | 1300-1600 |
| Bending | C=CH2 | 890 |
| Bending | HC=CHcis | 690 |
| Bending | HC=CHtrans | 965 |
| Bending | C=CHtrisubstitué | 790-840 |
la masse des atomes attachés à la liaison(plus les atomes sont gros plus faible
est la frequence)
la force de cette liaison. (plus forte est la constante plus élevée est la fréquence)
Il faut plus d'énergie pour étirer une liaison que de la déformer,donc
les bandes dues au stretching ont lieu à des fréquences plus élevées que
celles dues au bending.
L'intensité de la bande dépend de
l'existence de plusieurs groupes du même type
la valeur du moment dipolaire du groupe fonctionnel
la variation du moment dipolaire .
Plus il y a de groupes du même type et plus la liaison est polarisée,plus forte sera l'intensité.
2)La simulation du spectre theorique
2.a)La définition des degrés de liberté
Translation
La molécule peut se translater dans l'espace .
L'énergie cinétique de translation est définie par
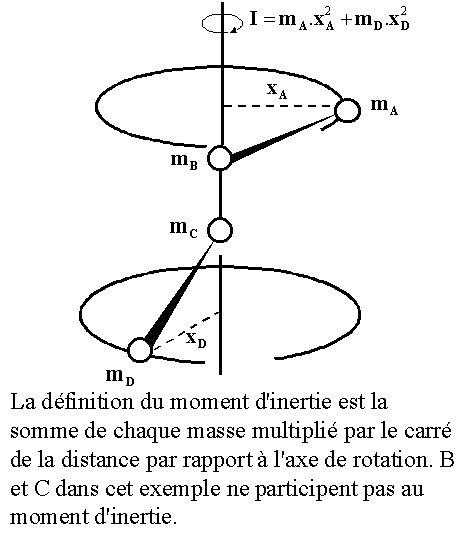
Rotation
La molécule peut également tourner autour de ses axes d'inertie.L'énergie cinétique
de rotation de la molécule est définie par
ou Ix, Iy, and Iz sont les moments d'inertie suivant les axes x, y, et z et wx,wy, etwz sont les vitesses angulaires autour de ces axes. La molécule dispose donc de 3 degrés de liberté en rotation(vx,vy,vz). (sauf dans le cas d'une molécule linéaire ou l'un des axes se confond avec les liaisons et le moment d'inertie correspondant est nul)
Vibration
Le nombre de degrés de liberté est de 3* N (n étant le nombre
total d'atomes)
Donc,en régle général,le nombre de modes de vibration est de
3*N- 6 pour une molécule polyatomique non linéaire
3*N- 5 pour une molécule polyatomique linéaire
Exemple
La molécule d'eau n’est pas linéaire, elle présente 3 modes de vibrations.
Le spectre observé donne, en effet trois bandes à 3756 cm-1, 3652 cm-1,
et 1545 cm-1.
| Vibration(cm-1) | N°1 | N°2 | N°3 |
| Expérimentale | 3756 | 3652 | 1545 |
| Theorique(Pm3) | 3991 | 3870 | 1742 |
2.b)La loi de Hooke
A l'équilibre,la fréquence n est définie par la relation suivante: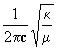
m = (m1*m2)/(m1 + m2)
I,Le moment d'inertie s’exprime alors simplement en fonction masse réduite par la relation
 Plus la force de liaison est grande plus la fréquence d'absorption est
élevée .Par exemple on s'attend a ce que la liaison acétylenique (C=-C )
ait une fréquence plus élevée que la liaison double ,celle ci étant plus
élevée que la liaison simple.
On peut dire en première approximation que:
Ks3= 3*Ks1
Ks2= 2*Ks1
Plus les atomes attachés à la liaison sont gros, plus la fréquence
de vibration est basse.Par exemple une liaison simple C-H devrait avoir
une fréquence plus élevée qu'une liaison C-C.
Plus la force de liaison est grande plus la fréquence d'absorption est
élevée .Par exemple on s'attend a ce que la liaison acétylenique (C=-C )
ait une fréquence plus élevée que la liaison double ,celle ci étant plus
élevée que la liaison simple.
On peut dire en première approximation que:
Ks3= 3*Ks1
Ks2= 2*Ks1
Plus les atomes attachés à la liaison sont gros, plus la fréquence
de vibration est basse.Par exemple une liaison simple C-H devrait avoir
une fréquence plus élevée qu'une liaison C-C.
2.c)La loi de Planck
.2.d)Calcul analytique des spectres IR
A partir d'une structure minimisée (en Mécanique Moléculaire,Semiempirique ou Abinitio), on calcule analytiquement la matrice des dérivées secondes(DE2/Dx2,DE2/Dy2, DE2/Dz2,..DE2/Dxy,DE2/Dxz... qui contient donc des sous-blocks de 3*3 éléments par atome. (Dés que le nombre d'atomes dans la molécule dépasse 50 ,le temps de calcul et l'espace mémoire deviennent prohibitifs.) Chaque élément de la matrice est ensuite réduit de la masse réduite correspondante, puis le programme extrait les fonctions et vecteurs propres. Des fréquences négatives nous indiquent que la molécule est mal minimisée ou qu'elle se trouve dans un minimum local trés éloigné de l'optimum global.2.e)Calcul des constantes de force à partir d'un spectre expérimental
Ex : Pour la laison C=O, on a M1 = 12 M2 = 16 Donc le poids de chaque atome en gramme est m1 =12/N m2 =16/N et m =1,03510-24 g r = 1,21 Ang; = 1,21 . 10-8cm I = m.r2=1.515.10-39 g.cm2| Molécule | C=-O | N=-O | HF | HCl | HBr | HI |
| Bande absorbée(cm-1) | 2143 | 1876 | 2905 | 2886 | 2559 | 2230 |
| Cte de force(10**5dyne.cm-1) | 18.4 | 15.3 | 9.7 | 4.8 | 4.1 | 3.2 |
