La recherche de conformations
La minimisation,"en une passe" donne parfois des hydrogénes
et des doublets disposés autour de l'atome dans des
positions structurales impossibles.
Ceci est souvent dû à un mouvement initial inapproprié de
ces atomes légers quand une structure fortement distordue
est introduite.
Celà peut également se produire si on démarre avec une
structure planaire.
C'est pourquoi le programme effectue une "seconde passe"
de minimisation aprés avoir repositionné les atomes
légers.
Une autre difficulté de la minimisation concerne le
probléme du minimum local.Les routines d'optimisation
sous contrainte ont en effet la fâcheuse tendance de
trouver un minimum d'energie le plus proche de la
structure d'entrée.En général,les distances et les angles
de liaison sont correctement minimisés,si bien que le
probléme du minima local peut se résumer à l'optimisation
des angles diédres(il faut beaucoup plus d'énergie
pour déformer une liaison ou un angle de valence par
rapport à un angle diédre).
Les approches suivantes ont pour but d'extraire la
molécule de son puits de potentiel.
1)La rotation des diédres
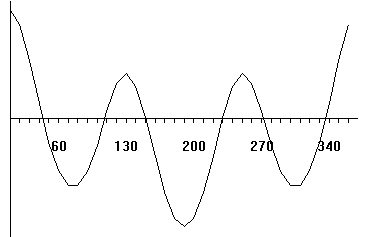
Butane:calcul des barriéres de rotation et affichage de la courbe E=f(angle diédre de la liaison centrale)
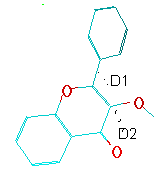

3-Methoxyflavone;Calcul de e=f(d1,d2) et affichage de l'isosurface


Webmaster:
Last
revised:05/2000